







































tumorigenic viruses
Tumorigenic viruses are subdivided according to the nucleic acid of their genome: Transforming retroviruses carry oncogenes originally derived from cellular genes that are involved in mitogenic signalling and growth control. DNA tumor viruses encode viral oncogenes that are essential for viral replication and cell transformation; viral oncoproteins complex with cellular proteins to stimulate cell cycle progression. This mechanism led to the discovery of tumor suppressors. Viral systems support the concept that cancer development occurs by the accumulation of multiple cooperating events.
It is estimated that 15% of all human tumors worldwide are caused by viruses. The infectious nature of viruses distinguishes them from all other carcinogenic factors. Tumor viruses primarily establish long-term persistent infections in humans, and carcinogenesis is an accidental side effect of viral replication strategies. Viruses are typically not complete carcinogens, with the known human cancer viruses displaying different roles in transformation. Many years usually pass between initial viral infection and tumor development, and most infected individuals do not develop cancer, although immunocompromised individuals are at elevated risk of viral-associated cancers.[ffta]
a. RNA genomes - hepatitis C virus (Flaviviridae, hepatocellular carcinoma (HCC)) retroviruses – human T-cell leukemia viruses (HTLVs), human immunodeficiency virus (HIV, Kaposi's sarcoma, acquired immunodeficiency syndrome).
b. DNA genomes - human papillomavirus (HPV, cervical cancer), adenoviruses, Epstein–Barr virus (nasopharyngeal carcinoma, Burkitt's lymphoma, post-transplant lymphomas, Hodgkin's disease) hepatitis B virus (Hepadnaviridae, hepatocellular carcinoma (HCC)),
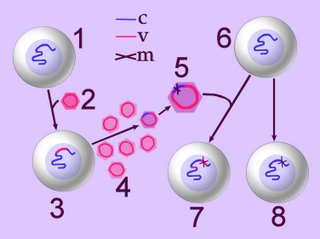
 Right - click to enlarge image – retroviral infection and oncogenesis. When a normal cell (1) is infected by a retrovirus (2), the viral reverse transcriptase reverse-transcribes the viral RNA into 'viral' DNA (v), which an integrase randomly integrates (inserts) into the host cell's genome (3-c-v). New viral particles are produced and shed by the infected cell (4) and some of these may contain proto-oncogene fragments of the host's genome (purple virion). Often, the transducted sequence undergoes mutation (m) into an oncogene (5) that is subsequently integrated into the genome of a second normal cell (6), which becomes transformed into a tumorigenic line (7). Under the influence of other carcinogens, normal cells may suffer mutation (m) of a proto-oncogene to an oncogene (8).
Right - click to enlarge image – retroviral infection and oncogenesis. When a normal cell (1) is infected by a retrovirus (2), the viral reverse transcriptase reverse-transcribes the viral RNA into 'viral' DNA (v), which an integrase randomly integrates (inserts) into the host cell's genome (3-c-v). New viral particles are produced and shed by the infected cell (4) and some of these may contain proto-oncogene fragments of the host's genome (purple virion). Often, the transducted sequence undergoes mutation (m) into an oncogene (5) that is subsequently integrated into the genome of a second normal cell (6), which becomes transformed into a tumorigenic line (7). Under the influence of other carcinogens, normal cells may suffer mutation (m) of a proto-oncogene to an oncogene (8).
Mechanisms of retroviral carcinogenesis:
First mechanism: Powerful transcriptional promoter sequences are located at the termini (ends) of the retroviral genome. These sequences are the 'long terminal repeats' (LTRs) that promote the transcription of the viral DNA into new virus particles.
Sometimes, in a process termed transduction, the process of integration causes rearrangement of the viral genome by incorporation of a portion of the host's genome into the viral genome. Occasionally, transduction provides the virus with a host gene that is normally involved in cell cycle control. The gene acquired from the host may be altered during the transduction process, in addition to its being transcribed at a higher rate by virtue of its association with the retroviral LTRs. In such cases, the transduced gene confers a growth advantage to the infected cell, causing the unrestricted cellular proliferation characteristic of tumorigenesis. These transduced host-cell-cycle genes function as oncogenes. The host gene that has been transduced is normally a cellular gene that functions as a proto-oncogene in its unmodified, non-transduced form. The genomes of transforming retroviruses contain numerous oncogenes.
Second mechanism: Long terminal repeats possess powerful transcription promoting effects. Retroviral genome integration into the host genome occurs randomly. Sometimes this integration process places the LTRs close to a gene that codes for a growth regulating protein. Abnormally elevated levels of expresson of such proteins can induce cellular transformation – 'retroviral integration induced transformation'. HIV induces certain forms of cancers by this integration induced transformation process.
In most cases, cellular transformation by DNA tumor viruses result from viral protein-host protein interaction. Proteins encoded by the DNA tumor viruses are termed tumor antigens or T antigens, and they can interact with cellular proteins. The interaction of T antigens with cellular proteins sequesters the cellular proteins away from their normal functional locations within the cell. Proteins sequestered by viral T antigens are predominantly tumor suppressor proteins, and the loss of their normal suppressor functions results in cellular transformation.
Human T-cell leukemia virus type-1 (HTLV-I) has been implicated with the etiology of adult T-cell leukemia (ATL) and certain other clinical disorders. Although the leukemogenic mechanism of HTLV-1 is not fully understood yet, the viral Tax protein is widely regarded as a key factor in this mechanism. Tax can modulate the synthesis or function of many regulatory factors which control a wide range of normal and oncogenic cellular processes and therefore, it acts as a potent oncoprotein. In the last few years, special attention has been attracted to Tax interference with the transactivation function of p53, a tumor suppressor protein that is involved in regulation of the cell-cycle and apoptosis and in maintaining the cellular genome integrity. p53 is mutated in about 60% of all human tumors. In contrast, mutant p53 is found in only small percentage of ATL patients. Nevertheless, p53 is inactive in the leukemic cells of most ATL patients and in most HTLV-1 transformed cells. By inactivating p53, Tax can immortalize the HTLV-1 infected cells and destabilize their genome. Consequently, such cells can progress towards the ultimate leukemic state by a stepwise accumulation of oncogenic mutations and other types of chromosomal aberrations. Furthermore, since p53 exists in most ATL patients in its wild type form, its reactivation by therapeutic drugs might be an effective approach for ATL therapy. Several mechanisms have been proposed so far for Tax-induced p53 inactivation. Tabakin-Fix Y, Azran I, Schavinky-Khrapunsky Y, Levy O, Aboud M. Functional inactivation of p53 by human T-cell leukemia virus type 1 Tax protein: mechanisms and clinical implications.
Carcinogenesis. 2006 Apr;27(4):673-81. Epub 2005 Nov 23.
Human Immunedeficiency Virus (HIV) is the cause of Acquired Immune Deficiency (AIDS). Although immunedeficiency syndromes were recognized prior to the discovery of HIV, the virus was discovered (isolated in '83) because an outbreak of unusual cases of Kaposi's sarcoma (since '81 in the San Francisco Bay Area).
Several viruses, including HIV/AIDS, control the expression of viral genes through viral binding sites for NF-κB, thus contributing to viral replication or viral pathogenicity. For HIV-1, activation of NF-κB could be related to activation of the virus from a latent, inactive state.
Many tumor types have chronically active NF-κB, resulting from:
● mutations in genes encoding the NF-κB transcription factors themselves, or
● mutations in genes that control NF-κB activity
Carcinogenesis of DNA viruses:
Human papillomavirus (HPV): HPV causes warts and is a risk factor for cancer of the cervix. HPV codes for the protein E6, which binds the tumor suppressor, p53 protein and inactivates it. This inactivation of p53, in synergy with the inactivation of another cell cycle regulator, p105RB, stimulates repeated cell division manifestested in HPV infection. HPV also encodes the E7 protein, which binds to the Rb protein preventing it from binding to the host transcription factor E2F. This frees E2F, which can now bind to the promoters of genes such as c-myc, stimulating the cell to enter the cell cycle.
Adenoviruses are used experimentally to induce carcinomas. Adenovirus remodels the cap-initiation complex required for translation of mRNAs. Adenovirus simultaneously inhibits cap-dependent host cell mRNA translation while promoting the translation of its late viral mRNAs during infection. Adenovirus cap-remodeling utilizes a viral protein that displaces a protein kinase from the initiation complex, and tyrosine kinase activity plays a central role in the control of late adenovirus protein synthesis. Adenovirus protein 100k blocks cellular mRNA translation by disrupting the cap-initiation complex and promotes viral mRNA translation through the alternate mechanism of ribosome shunting in a tyrosine phosphorylation-dependent manner. 100k protein interaction with initiation factor eIF4G and the viral 5' noncoding region on viral late mRNAs, known as the tripartite leader, are both essential for ribosome shunting. Heat shock induced cell stress uses heat shock proteins to inhibit cellular protein synthesis by mediating dissociation of the cap-initiation complex. Adenovirus and heat shock mRNAs utilize the altered cap-initiation complex to undergo the unusual form of translation initiation of ribosome shunting.
Hepatitis B virus (HBV) is a para-retrovirus that is associated with human liver cancer. The HBx protein is a regulatory protein that is involved in carcinogenesis and HBV infection. HBx activates Src-Ras signal transduction pathways, which are critical for HBx activation of many transcription factors, and for HBx activation of HBV reverse-transcription and DNA replication. HBx causes a loss of early cell-cycle checkpoint controls , it is probably involved in HBV pathogenesis by cytokines, and it may play a role in liver cancer caused by Hepatitis B virus infection.[s] image [] model for HBx []
The emerging p53 gene family.
Perturbation of p53 protein function is a common, if not universal, finding in human cancer. Tumor suppression by p53 is due, at least in part, to its ability to activate transcription of certain genes involved in cell cycle control and apoptosis (programmed cell death). Two additional members of the mammalian p53 family, p73 and p51, which is also known as p40, p63, KET, or p73L, were recently identified. Both of these proteins share substantial sequence homology with p53 and can, at least when overproduced, activate p53-responsive promoters and induce apoptosis. Nonetheless, data on differences between these proteins and p53 are emerging. For example, p73 is not induced by DNA damage and is not targeted for inactivation by viral oncoproteins such as simian virus 40 (SV40) T antigen, adenovirus E1B 55K, and human papillomavirus E6. In contrast to p53, neither p73 nor p51 appears to be frequently mutated in human cancers on the basis of the limited studies reported to date. Finally, unlike p53, cells produce multiple p73 and p51 isoforms as a result of alternative splicing, and production of p73 and p51 appears to be restricted to certain tissues. Additional studies are required to determine the role, if any, that p73 and p51 play in cell growth control and carcinogenesis. Kaelin WG Jr. The emerging p53 gene family. Free Full Text Article J Natl Cancer Inst. 1999 Apr 7;91(7):594-8.
¤ Cancer ¤ carcinogenesis ¤ oncogenes ¤ proliferation ¤ retroviruses ¤ signaling molecules ¤ tumor suppressors ¤ tumorigenic viruses ¤ Tables Malignant Transformation Oncogenes Proto-oncogenes
▲ Top ▲

It is estimated that 15% of all human tumors worldwide are caused by viruses. The infectious nature of viruses distinguishes them from all other carcinogenic factors. Tumor viruses primarily establish long-term persistent infections in humans, and carcinogenesis is an accidental side effect of viral replication strategies. Viruses are typically not complete carcinogens, with the known human cancer viruses displaying different roles in transformation. Many years usually pass between initial viral infection and tumor development, and most infected individuals do not develop cancer, although immunocompromised individuals are at elevated risk of viral-associated cancers.[ffta]
a. RNA genomes - hepatitis C virus (Flaviviridae, hepatocellular carcinoma (HCC)) retroviruses – human T-cell leukemia viruses (HTLVs), human immunodeficiency virus (HIV, Kaposi's sarcoma, acquired immunodeficiency syndrome).
b. DNA genomes - human papillomavirus (HPV, cervical cancer), adenoviruses, Epstein–Barr virus (nasopharyngeal carcinoma, Burkitt's lymphoma, post-transplant lymphomas, Hodgkin's disease) hepatitis B virus (Hepadnaviridae, hepatocellular carcinoma (HCC)),
{kind=link}
 Right - click to enlarge image – retroviral infection and oncogenesis. When a normal cell (1) is infected by a retrovirus (2), the viral reverse transcriptase reverse-transcribes the viral RNA into 'viral' DNA (v), which an integrase randomly integrates (inserts) into the host cell's genome (3-c-v). New viral particles are produced and shed by the infected cell (4) and some of these may contain proto-oncogene fragments of the host's genome (purple virion). Often, the transducted sequence undergoes mutation (m) into an oncogene (5) that is subsequently integrated into the genome of a second normal cell (6), which becomes transformed into a tumorigenic line (7). Under the influence of other carcinogens, normal cells may suffer mutation (m) of a proto-oncogene to an oncogene (8).
Right - click to enlarge image – retroviral infection and oncogenesis. When a normal cell (1) is infected by a retrovirus (2), the viral reverse transcriptase reverse-transcribes the viral RNA into 'viral' DNA (v), which an integrase randomly integrates (inserts) into the host cell's genome (3-c-v). New viral particles are produced and shed by the infected cell (4) and some of these may contain proto-oncogene fragments of the host's genome (purple virion). Often, the transducted sequence undergoes mutation (m) into an oncogene (5) that is subsequently integrated into the genome of a second normal cell (6), which becomes transformed into a tumorigenic line (7). Under the influence of other carcinogens, normal cells may suffer mutation (m) of a proto-oncogene to an oncogene (8).Mechanisms of retroviral carcinogenesis:
First mechanism: Powerful transcriptional promoter sequences are located at the termini (ends) of the retroviral genome. These sequences are the 'long terminal repeats' (LTRs) that promote the transcription of the viral DNA into new virus particles.
Sometimes, in a process termed transduction, the process of integration causes rearrangement of the viral genome by incorporation of a portion of the host's genome into the viral genome. Occasionally, transduction provides the virus with a host gene that is normally involved in cell cycle control. The gene acquired from the host may be altered during the transduction process, in addition to its being transcribed at a higher rate by virtue of its association with the retroviral LTRs. In such cases, the transduced gene confers a growth advantage to the infected cell, causing the unrestricted cellular proliferation characteristic of tumorigenesis. These transduced host-cell-cycle genes function as oncogenes. The host gene that has been transduced is normally a cellular gene that functions as a proto-oncogene in its unmodified, non-transduced form. The genomes of transforming retroviruses contain numerous oncogenes.
Second mechanism: Long terminal repeats possess powerful transcription promoting effects. Retroviral genome integration into the host genome occurs randomly. Sometimes this integration process places the LTRs close to a gene that codes for a growth regulating protein. Abnormally elevated levels of expresson of such proteins can induce cellular transformation – 'retroviral integration induced transformation'. HIV induces certain forms of cancers by this integration induced transformation process.
In most cases, cellular transformation by DNA tumor viruses result from viral protein-host protein interaction. Proteins encoded by the DNA tumor viruses are termed tumor antigens or T antigens, and they can interact with cellular proteins. The interaction of T antigens with cellular proteins sequesters the cellular proteins away from their normal functional locations within the cell. Proteins sequestered by viral T antigens are predominantly tumor suppressor proteins, and the loss of their normal suppressor functions results in cellular transformation.
Human T-cell leukemia virus type-1 (HTLV-I) has been implicated with the etiology of adult T-cell leukemia (ATL) and certain other clinical disorders. Although the leukemogenic mechanism of HTLV-1 is not fully understood yet, the viral Tax protein is widely regarded as a key factor in this mechanism. Tax can modulate the synthesis or function of many regulatory factors which control a wide range of normal and oncogenic cellular processes and therefore, it acts as a potent oncoprotein. In the last few years, special attention has been attracted to Tax interference with the transactivation function of p53, a tumor suppressor protein that is involved in regulation of the cell-cycle and apoptosis and in maintaining the cellular genome integrity. p53 is mutated in about 60% of all human tumors. In contrast, mutant p53 is found in only small percentage of ATL patients. Nevertheless, p53 is inactive in the leukemic cells of most ATL patients and in most HTLV-1 transformed cells. By inactivating p53, Tax can immortalize the HTLV-1 infected cells and destabilize their genome. Consequently, such cells can progress towards the ultimate leukemic state by a stepwise accumulation of oncogenic mutations and other types of chromosomal aberrations. Furthermore, since p53 exists in most ATL patients in its wild type form, its reactivation by therapeutic drugs might be an effective approach for ATL therapy. Several mechanisms have been proposed so far for Tax-induced p53 inactivation. Tabakin-Fix Y, Azran I, Schavinky-Khrapunsky Y, Levy O, Aboud M. Functional inactivation of p53 by human T-cell leukemia virus type 1 Tax protein: mechanisms and clinical implications.
Carcinogenesis. 2006 Apr;27(4):673-81. Epub 2005 Nov 23.
Human Immunedeficiency Virus (HIV) is the cause of Acquired Immune Deficiency (AIDS). Although immunedeficiency syndromes were recognized prior to the discovery of HIV, the virus was discovered (isolated in '83) because an outbreak of unusual cases of Kaposi's sarcoma (since '81 in the San Francisco Bay Area).
Several viruses, including HIV/AIDS, control the expression of viral genes through viral binding sites for NF-κB, thus contributing to viral replication or viral pathogenicity. For HIV-1, activation of NF-κB could be related to activation of the virus from a latent, inactive state.
Many tumor types have chronically active NF-κB, resulting from:
● mutations in genes encoding the NF-κB transcription factors themselves, or
● mutations in genes that control NF-κB activity
Carcinogenesis of DNA viruses:
Human papillomavirus (HPV): HPV causes warts and is a risk factor for cancer of the cervix. HPV codes for the protein E6, which binds the tumor suppressor, p53 protein and inactivates it. This inactivation of p53, in synergy with the inactivation of another cell cycle regulator, p105RB, stimulates repeated cell division manifestested in HPV infection. HPV also encodes the E7 protein, which binds to the Rb protein preventing it from binding to the host transcription factor E2F. This frees E2F, which can now bind to the promoters of genes such as c-myc, stimulating the cell to enter the cell cycle.
Adenoviruses are used experimentally to induce carcinomas. Adenovirus remodels the cap-initiation complex required for translation of mRNAs. Adenovirus simultaneously inhibits cap-dependent host cell mRNA translation while promoting the translation of its late viral mRNAs during infection. Adenovirus cap-remodeling utilizes a viral protein that displaces a protein kinase from the initiation complex, and tyrosine kinase activity plays a central role in the control of late adenovirus protein synthesis. Adenovirus protein 100k blocks cellular mRNA translation by disrupting the cap-initiation complex and promotes viral mRNA translation through the alternate mechanism of ribosome shunting in a tyrosine phosphorylation-dependent manner. 100k protein interaction with initiation factor eIF4G and the viral 5' noncoding region on viral late mRNAs, known as the tripartite leader, are both essential for ribosome shunting. Heat shock induced cell stress uses heat shock proteins to inhibit cellular protein synthesis by mediating dissociation of the cap-initiation complex. Adenovirus and heat shock mRNAs utilize the altered cap-initiation complex to undergo the unusual form of translation initiation of ribosome shunting.
Hepatitis B virus (HBV) is a para-retrovirus that is associated with human liver cancer. The HBx protein is a regulatory protein that is involved in carcinogenesis and HBV infection. HBx activates Src-Ras signal transduction pathways, which are critical for HBx activation of many transcription factors, and for HBx activation of HBV reverse-transcription and DNA replication. HBx causes a loss of early cell-cycle checkpoint controls , it is probably involved in HBV pathogenesis by cytokines, and it may play a role in liver cancer caused by Hepatitis B virus infection.[s] image [] model for HBx []
The emerging p53 gene family.
Perturbation of p53 protein function is a common, if not universal, finding in human cancer. Tumor suppression by p53 is due, at least in part, to its ability to activate transcription of certain genes involved in cell cycle control and apoptosis (programmed cell death). Two additional members of the mammalian p53 family, p73 and p51, which is also known as p40, p63, KET, or p73L, were recently identified. Both of these proteins share substantial sequence homology with p53 and can, at least when overproduced, activate p53-responsive promoters and induce apoptosis. Nonetheless, data on differences between these proteins and p53 are emerging. For example, p73 is not induced by DNA damage and is not targeted for inactivation by viral oncoproteins such as simian virus 40 (SV40) T antigen, adenovirus E1B 55K, and human papillomavirus E6. In contrast to p53, neither p73 nor p51 appears to be frequently mutated in human cancers on the basis of the limited studies reported to date. Finally, unlike p53, cells produce multiple p73 and p51 isoforms as a result of alternative splicing, and production of p73 and p51 appears to be restricted to certain tissues. Additional studies are required to determine the role, if any, that p73 and p51 play in cell growth control and carcinogenesis. Kaelin WG Jr. The emerging p53 gene family. Free Full Text Article J Natl Cancer Inst. 1999 Apr 7;91(7):594-8.
¤ Cancer ¤ carcinogenesis ¤ oncogenes ¤ proliferation ¤ retroviruses ¤ signaling molecules ¤ tumor suppressors ¤ tumorigenic viruses ¤ Tables Malignant Transformation Oncogenes Proto-oncogenes
▲ Top ▲
Labels: growth control, HCC, HIV, HPV, HTLV, LTR, mitogenic signalling, oncogene, p53, proto-oncogene, retrovirus, reverse transcriptase, tumor suppressor, tumorigenic viruses